
Research
Living organisms sustain tissues and entire bodies through repeated cycles of cell division, the smallest functional unit of life. During each division, DNA—the carrier of genomic information—must be accurately duplicated within a limited timeframe. However, DNA is an enormous molecule that adopts diverse structures, and its replication is far from a straightforward process. Moreover, DNA replication does not proceed indefinitely with uniform fidelity. As cells age, the risk of replication failure increases, elevating the likelihood of cell death and genomic instability, including mutations and chromosomal abnormalities caused by defects in the replication machinery. Alterations in genetic information arising from DNA replication generate diversity within biological populations and serve as a driving force of evolution. In multicellular organisms, including humans, however, such changes can impair tissue and organ function and give rise to cell populations that deviate from physiological homeostasis, such as cancer cells. Cells contain a wide variety of DNA polymerases with distinct efficiencies and fidelities in DNA synthesis. Elucidating their respective roles in genome replication is essential for understanding the mechanisms that maintain genomic stability. Our research focuses on uncovering how DNA polymerases with diverse properties cooperate to execute genome replication and on defining the intrinsic flexibility embedded within the replication machinery. To achieve this, we combine detailed analyses of individual molecular components required for DNA replication with genome-wide experimental approaches supported by computational methods to generate comprehensive replication profiles across the entire genome. Furthermore, we investigate how DNA replication programmes are altered during cancer evolution, with the goal of establishing a foundation for novel strategies in cancer prevention and therapy.
Current Projects
- Coordinated functions of DNA polymerases during genome replication We have developed a technique termed Polymerase Usage Sequencing (Pu-seq), which enables genome-wide analysis of the synthesis tracts of individual DNA polymerases, and we are applying this approach to elucidate their roles in DNA replication. Using this method, we have clarified the division of labour between DNA polymerases responsible for leading- and lagging-strand synthesis at the replication fork. We are now extending this technology beyond the three major replicative DNA polymerases with substantial contributions to genome duplication (Polδ, Polε and Polα) to include a broader range of error-prone DNA polymerases, with the aim of defining how polymerase function influences mutagenesis. In this context, we integrate our analyses with cancer genome mutation data from public databases, including The Cancer Genome Atlas (TCGA), to elucidate the mechanisms underlying mutation generation in tumours and to identify replication vulnerabilities that represent critical weak points in individual cancer types.
- Changes in DNA replication function in cancer cells and their impact During cancer progression, oncogenic activation of drivers such as Ras and c-Myc accelerates cell-cycle progression and induces high-level expression of genes involved in proliferation. Under these conditions, genome replication can be initiated prematurely, before a full complement of replication factors has been properly assembled on chromosomal DNA. As a consequence, a reduction in replication initiation sites and related alterations can generate genomic regions that are difficult for the replication machinery to traverse. Although a link between such dramatic changes in replication dynamics and elevated mutation rates has long been discussed, the overall picture remains largely unresolved. We aim to define how oncogene expression reshapes DNA polymerase function and the mechanisms governing replication initiation, progression and termination, thereby clarifying the inherent flexibility and instability of the replication programme.
- Mechanisms by which transcription machinery interferes with DNA replication In eukaryotic cells, multiple replication origins are distributed along each chromosome, making it impossible to completely avoid collisions or interference between replication forks and the transcription machinery. This risk is further increased in mammalian cells, which harbour numerous large genes exceeding 100 kb and containing long introns, thereby heightening the likelihood of replication–transcription conflicts. It has been proposed that such conflicts are mitigated by temporal separation of transcription and replication during different phases of the cell cycle, as well as by their spatial segregation within the nucleus. However, the extent to which these mechanisms operate in vivo remains unclear. As illustrated in Figure 1, we quantitatively and precisely evaluate replication fork initiation and termination sites, as well as fork directionality across the genome, by analysing the synthesis profiles of DNA polymerases involved in genome replication. Using this approach, we aim to determine how gene transcription, including transcription within non-coding regions, influences replication fork progression and structure. Furthermore, we investigate the factors involved in the formation and resolution of transcription–replication conflicts (TRCs) and define the features of TRCs that pose a high risk of inducing chromosomal instability.
- Effects of replication stress on the tumor microenvironment The DNA replication machinery is frequently challenged by various sources of stress, leading to local reductions in replication efficiency as well as replication fork stalling and collapse. These events not only compromise the efficiency of genome duplication but also generate processed DNA intermediates that can leak into the cytoplasm. Cytosolic DNA is sensed by cyclic GMP-AMP synthase (cGAS) and activates the STimulator of INterferon Genes (STING) pathway, triggering intracellular inflammatory signaling. As a consequence, inflammatory responses are induced within the affected cell and can also influence neighboring cells, thereby altering cellular proliferative capacity and tissue behavior. Replication stress–induced inflammation can therefore impact not only tumor cells but also the surrounding tissues and microenvironment, exerting either tumor-promoting or tumor-suppressive effects depending on the context. Building on efforts to elucidate the mechanisms by which replication stress provokes inflammatory responses, we aim to explore strategies to control tumor growth by regulating cytoplasmic DNA derived from perturbed replication processes.
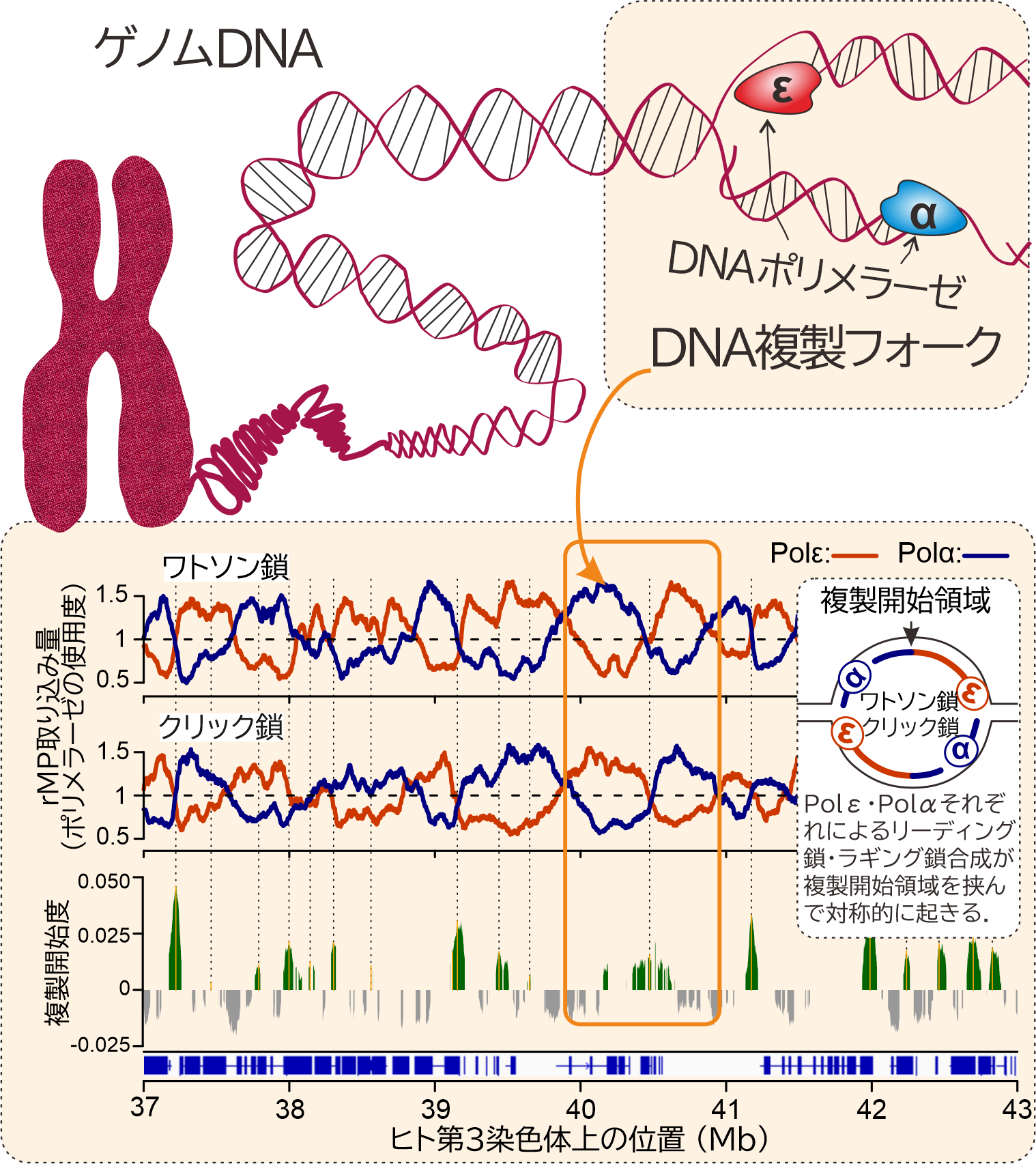 Figure 1: Genome-wide analysis of DNA polymerase functions
Figure 1: Genome-wide analysis of DNA polymerase functions
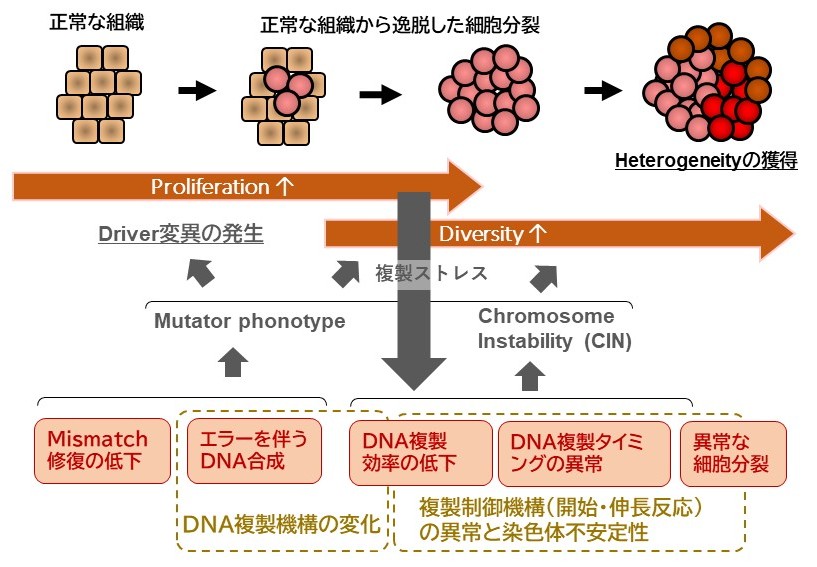 Figure 2: Changes in replication function during cancer progression
Figure 2: Changes in replication function during cancer progression
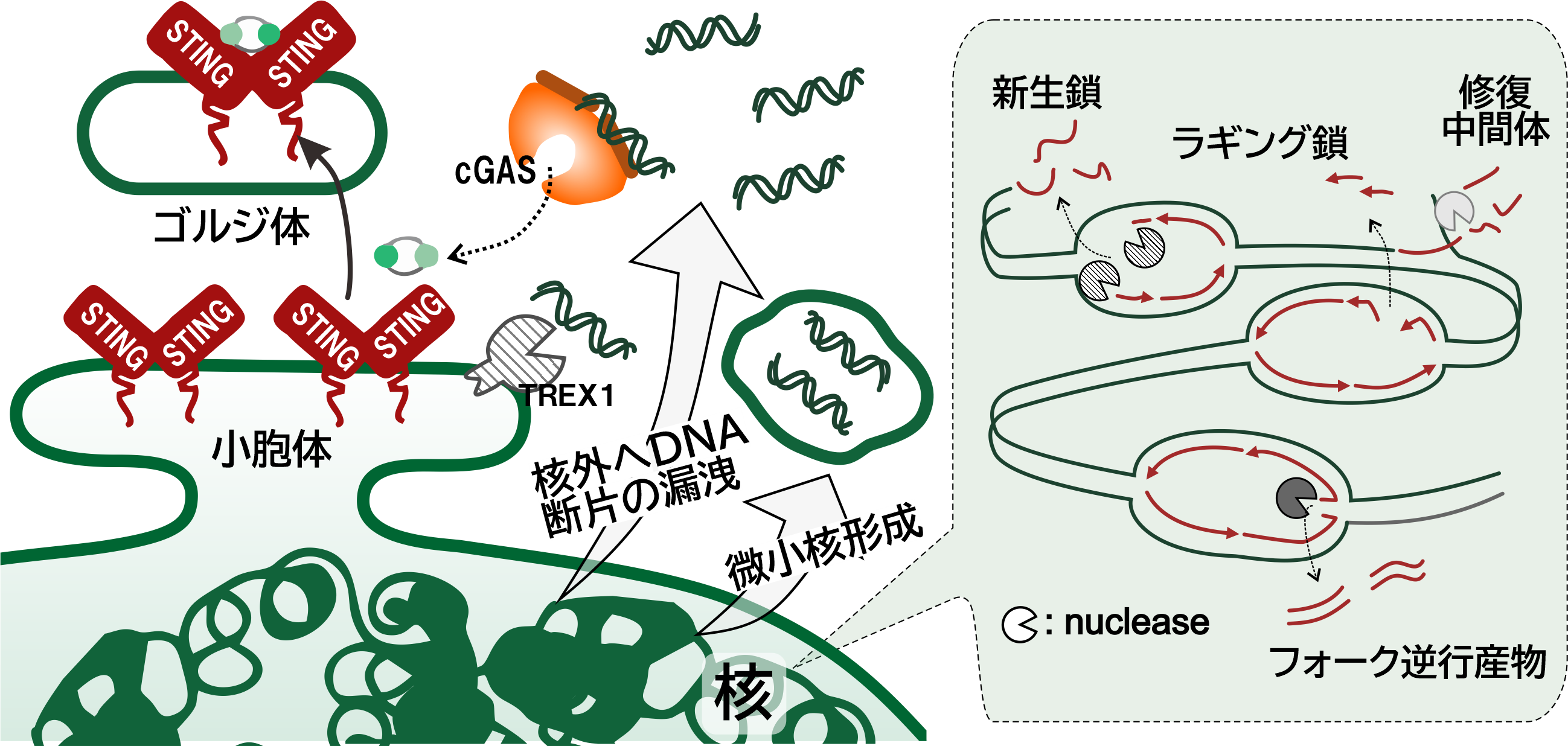 Figure 3: Inflammatory response induced by replication stress
Figure 3: Inflammatory response induced by replication stress