

![]()
- HOME
- Outstanding Progress

Cytotoxic activity of Tivantinib (ARQ 197) is not due solely to MET inhibition
Ryohei Katayama1,2,3, Aki Aoyama3,5, Takao Yamori4,6, Jie Qi1, Tomoko Oh-hara3, Youngchul Song1, Jeffrey A. Engelman1,2, and Naoya Fujita1,3
1Massachusetts General Hospital Cancer Center; 2Department of Medicine, Harvard Medical School, Boston, Massachusetts; Divisions of 3Experimental Chemotherapy and 4Molecular Pharmacology, Cancer Chemotherapy Center, Japanese foundation for Cancer Research; 5Department of Medical Genome Sciences, Graduate School of Frontier Sciences, The University of Tokyo; and 6Pharmaceuticals and Medical Devices Agency (PMDA), Tokyo, Japan
R. Katayama and A. Aoyama contributed equally to this work.
Cancer Research, in press
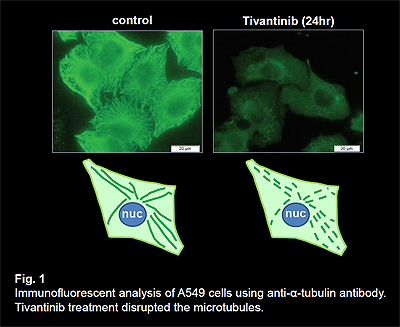 The receptor tyrosine kinase c-MET is the high-affinity receptor for the hepatocyte growth factor (HGF). The HGF/c-MET axis is often dysregulated in tumors. c-MET activation can be caused by MET gene amplification, activating mutations, and auto- or paracrine mechanisms. Thus, c-MET inhibitors are under development as anti-cancer drugs. Tivantinib (ARQ 197) was reported as a small molecule c-MET inhibitor and early clinical studies suggest anti-tumor activity. To assess if the anti-tumor activity of tivantinib was due to inhibition of c-MET, we compared the activity of tivantinib to other c-MET inhibitors in both c-MET addicted and non-addicted cancer cells. As expected, other c-MET inhibitors, crizotinib and PHA-665752, suppressed the growth of c-MET addicted cancers, but not the growth of cancers that are not addicted to c-MET. In contrast, tivantinib inhibited cell viability with similar potency in both c-MET addicted and non-addicted cells. These results suggest that tivantinib exhibits its antitumor activity in a manner independent of c-MET status. Tivantinib treatment induced a G2/M cell cycle arrest in EBC1 cells similarly to vincristine treatment, whereas PHA-665752 or crizotinib treatment markedly induced G0/G1 cell cycle arrest. To identify the additional molecular target of tivantinib, we performed COMPARE analysis , an in silico screening of a database of drug sensitivities across 39 cancer cell lines (JFCR39), and identified microtubule as a target of tivantinib. Tivantinib treated cells demonstrated typical microtubule disruption similar to vincristine (Fig. 1) and inhibited microtubule assembly in vitro (Fig. 2).
The receptor tyrosine kinase c-MET is the high-affinity receptor for the hepatocyte growth factor (HGF). The HGF/c-MET axis is often dysregulated in tumors. c-MET activation can be caused by MET gene amplification, activating mutations, and auto- or paracrine mechanisms. Thus, c-MET inhibitors are under development as anti-cancer drugs. Tivantinib (ARQ 197) was reported as a small molecule c-MET inhibitor and early clinical studies suggest anti-tumor activity. To assess if the anti-tumor activity of tivantinib was due to inhibition of c-MET, we compared the activity of tivantinib to other c-MET inhibitors in both c-MET addicted and non-addicted cancer cells. As expected, other c-MET inhibitors, crizotinib and PHA-665752, suppressed the growth of c-MET addicted cancers, but not the growth of cancers that are not addicted to c-MET. In contrast, tivantinib inhibited cell viability with similar potency in both c-MET addicted and non-addicted cells. These results suggest that tivantinib exhibits its antitumor activity in a manner independent of c-MET status. Tivantinib treatment induced a G2/M cell cycle arrest in EBC1 cells similarly to vincristine treatment, whereas PHA-665752 or crizotinib treatment markedly induced G0/G1 cell cycle arrest. To identify the additional molecular target of tivantinib, we performed COMPARE analysis , an in silico screening of a database of drug sensitivities across 39 cancer cell lines (JFCR39), and identified microtubule as a target of tivantinib. Tivantinib treated cells demonstrated typical microtubule disruption similar to vincristine (Fig. 1) and inhibited microtubule assembly in vitro (Fig. 2).
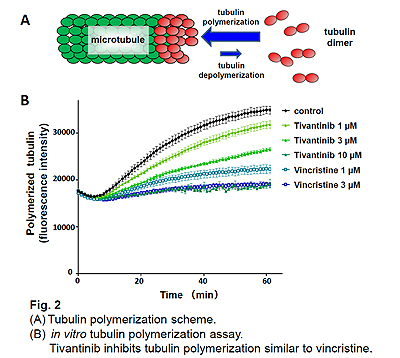 These results suggest that tivantinib inhibits microtubule polymerization in addition to inhibiting c-MET. Many clinical trials were based on the fact that tivantinib was a c-MET inhibitor. The phase II trial of erlotinib with tivantinib or placebo did not reveal a significant difference between 2 groups. Because our study suggests that tivantinib exhibits antitumor effect via microtubule inhibition, it remains possible that tivantinib may also be effective to other solid cancers. Further studies are necessary to understand the detailed mechanism how tivantinib inhibits tubulin polymerization and how to use this agent most effectively to treat cancer.
These results suggest that tivantinib inhibits microtubule polymerization in addition to inhibiting c-MET. Many clinical trials were based on the fact that tivantinib was a c-MET inhibitor. The phase II trial of erlotinib with tivantinib or placebo did not reveal a significant difference between 2 groups. Because our study suggests that tivantinib exhibits antitumor effect via microtubule inhibition, it remains possible that tivantinib may also be effective to other solid cancers. Further studies are necessary to understand the detailed mechanism how tivantinib inhibits tubulin polymerization and how to use this agent most effectively to treat cancer.
![]()

