




【プレスリリース】 白血病細胞の骨髄への定着に必要な新たな遺伝子を発見
2016年03月29日
ポイント
l 白血病細胞が骨髄に定着し根を下ろすことは、治療を困難にする要因の一つですが、その原因となる遺伝子Sytl1の同定に成功しました。
l Sytl1が活性化されると、白血病細胞の中に存在するシグナル因子の輸送が盛んになり、白血病細胞と骨髄内の間質細胞の結びつきが促進されます。
l 今回発見されたSytl1の活性化は、生体内での白血病細胞の定着機構を説明する全く新しい現象で、この分子回路を標的とした治療薬の開発につながることが期待されます。
横山隆志 研究員(がん研究会がん研究所発がん研究部)と 中村卓郎 部長(がん研究会がん研究所発がん研究部)、及び東京大学、東京理科大学、エラスムス大学の研究グループは、急性骨髄性白血病のモデルマウスを用いて、白血病細胞が骨髄に定着する仕組みを解明しました。
今回、若年者・高齢者ともに発症する急性骨髄性白血病で、原因遺伝子の一つであるMeis1が白血病細胞の骨髄定着の鍵を握っていることがわかりました。血液を作る場所である骨髄には、骨髄ニッチと呼ばれる微小環境が重要です。白血病細胞は、しばしばこの骨髄ニッチを占拠して、正常な血液細胞の形成を妨げるだけではなく、骨髄ニッチに潜むことで治療への抵抗力を増すことが知られています。Meis1は転写因子の一種で、特定の遺伝子(標的遺伝子)のタンパク産生につながるmRNA合成を調節する機能をもっています。しかし、白血病細胞の維持に重要なMeis1の標的遺伝子は、これまで良く分かっていませんでした。本研究グループは、白血病ゲノム中にあるMeis1タンパクの結合部位を解析し、多数の候補遺伝子の中からSytl1遺伝子が白血病細胞の骨髄定着に必要なことを見出しました。Meis1は、Sytl1を活性化することによって、白血病細胞の運動と骨髄ニッチとの相互作用、さらに生体内での骨髄への定着をコントロールしていることが分かりました。Sytl1の発現上昇はヒトの急性骨髄性白血病でも生じており、ヒト白血病細胞でもSytl1遺伝子の発現を抑えると、骨髄定着が抑制されることが分かりました。今後、Sytl1を分子標的として利用することで、新しい治療薬の開発や、白血病の治療抵抗性の要因となる白血病幹細胞を抑制する足掛かりになることが期待されます。
本研究の成果は、米国の医学雑誌『Journal of Clinical Investigation』オンライン版(米国時間3月28日付:日本時間3月29日)に掲載されました。
<研究の背景>
急性骨髄性白血病は、若年者、高齢者ともに発生が見られる血液のがんで、化学療法や骨髄移植の進歩により治療成績は向上しています。しかし、病型によって治療成績は異なり、悪性度が高く難治性の症例も少なくないこと、特に難治例では骨髄ニッチに潜む白血病幹細胞が治療抵抗性を左右することが問題となっています。体の中で白血病細胞が骨髄ニッチを初めとする微小環境とどのように関わって生存し、増殖するかはまだ良くわかっていません。本研究グループは急性骨髄性白血病のモデルマウスを使って、白血病細胞が骨髄ニッチに定着するために必要な分子Sytl1を新たに同定しました。この発見が白血病の新たな治療法の開発につながるものと考えています。
<研究の内容>
1)白血病細胞の骨髄内定着にはMeis1遺伝子が必要である
これまでの研究により、転写因子1)をコードするMeis1遺伝子は、急性骨髄性白血病の発症と正常造血に極めて重要な役割を担っていることがわかっています。例えば、Meis1のノックアウトマウスでは、造血幹細胞の長期間の維持が困難であったり、血小板が全く出来ないなど、造血に重篤な障害が生じます。Meis1は、同じくホメオボックス転写因子のHOXのパートナーとして組織発生や造血で機能し、白血病の発症においてもHoxa9遺伝子と協調作用を示します。 しかし、Meis1が白血病の発症や進展において、どのような現象に関わっているか、その具体的な機能は不明でした。一方、Hoxa9遺伝子は、造血細胞に対して強力な発がん作用を持っていて、造血幹細胞や前駆細胞を容易に不死化することが出来ますが、Meis1との協調作用がないとマウスの生体内で白血病を発症することは出来ません。本研究グループは、Hoxa9とMeis1両者が発現している白血病細胞を使って、Meis1をノックアウトすると白血病細胞の骨髄ニッチ2)に定着が劇的に阻止されることを見出しました。それだけではなく、Meis1が存在しないと、白血病細胞と骨髄の間質細胞との接着や、間質細胞が産生しているサイトカイン3)に対する遊走能も阻害されることが分かりました。つまり、Meis1は、白血病細胞が骨髄ニッチに定着する為に、ニッチに存在する間質細胞から何らかのシグナルを受け取って、間質細胞との相互作用を促進している可能性が示されました。
2)Meis1は標的遺伝子のSytl1を活性化して白血病細胞の骨髄内定着を促進している
Meis1は転写因子として働いているため、特異的な標的遺伝子を活性化して白血病細胞の骨髄内定着を促進していることが予測されました。そこで、本研究グループは、クロマチン免疫沈降法を使ってMeis1が白血病細胞のゲノム上で結合している部位を網羅的に解析して近傍の遺伝子を選択し、Meis1の有無で発現が上昇することや造血・がん化に関連する機能を持っていることなどの条件で絞り込みを行いました。次に、絞り込んだ有力候補の7遺伝子をHoxa9と一緒にマウスの造血細胞に導入して、白血病を発症させる能力の有無を調べました。その結果、Sytl1遺伝子がMeis1遺伝子の機能を肩代わりして、Hoxa9の白血病発症能を助けることが分かりました(図1)。それだけではなく、Sytl1は前項で示したMeis1による白血病細胞の骨髄ニッチへの定着作用、間質細胞との接着、サイトカインに対する遊走促進能全てについて、Meis1が存在しない際に機能を補完することが明らかとなりました。また、このような現象はSytl1のノックアウトマウスから採取した造血細胞では全く認められません。Meis1はSytl1のプロモーター領域4)に結合し、そのmRNA産生を増加させることから、Sytl1を直接の標的遺伝子として制御していることも分かりました。白血病の発症や進展においてMeis1が担っている機能の多くは、Sytl1を介して達成されていることが示された訳です。
3)Sytl1は細胞内の受容体CXCR4の膜輸送を促進してCXCL12に対する応答効果を上げ、白血病細胞の骨髄ニッチへの定着の鍵を握っている
Sytl1タンパクは細胞内に存在する小さな顆粒を膜に効率良く輸送する役割を担っています。これらの顆粒の中には、消化酵素や凝固因子、マイクロRNAといった数多くの生理活性物質が細胞の種類に応じて含まれています。その中で本研究グループは、CXCR4というケモカイン(サイトカインの1グループ)受容体に注目しました。骨髄ニッチの中で造血幹細胞の維持に非常に重要なCAR細胞と言われる間質細胞は、CXCL12というケモカインを分泌しており、CAR細胞のCXCL12が造血幹細胞のCXCR4に結合することにより発生するシグナルが、造血幹細胞の生存、自己複製、分化に必要であると考えられています。また、白血病細胞の増殖や維持にも重要である事実も知られています。Sytl1が活性化していると、CXCL12に対する白血病細胞の運動能が増加することが分かりました。CXCR4はCXCL12を受け取ってシグナルを伝えると速やかに破壊されるため、シグナルの動きは一過性ですが、Sytl1は新しいCXCR4を次々と細胞膜に送り込むことによって、より効率良くCXCL12を取り込むことが示されました(図1)。この現象は、細胞内のシグナルの動きとも一致していることも分かりました。
4)SYTL1はヒトの白血病でもしばしば発現が増加している
これまでに述べたMeis1やSytl1による白血病細胞の骨髄ニッチへの定着促進作用は、マウスの白血病モデルを使った実験で示されたものです。そこで、ヒトの急性骨髄性白血病526例におけるSytl1の発現を調べてみました。すると、Sytl1はMeis1やHoxa9の発現が高い症例で同時に発現が増加していることが示されました。Meis1やHoxa9はMLL遺伝子が異常を示す白血病グループで発現が高いことが知られていましたが、Sytl1の発現増加は、MLLグループの中でも特に予後不良な症例と相関することも分かりました。同時に、ヒトの白血病細胞を使った場合もSytl1がCXCL12の分子経路を介して骨髄ニッチへの定着促進に関わっていることも明らかになりました。今後Sytl1を新たな分子標的とする治療法を開発し、難治性の白血病の治療、特に化学療法に抵抗性を示す白血病幹細胞を根絶する治療の導入が期待されます。
<まとめ>
「今回の研究成果により、白血病の難治化の要因の一つである、白血病細胞の骨髄ニッチ定着を促進する仕組みが明らかになりました(図1)。その鍵となる分子Sytl1の役割が脚光を浴びたのは、白血病はもとより、がん全体としても初めてのことです。本研究の成果を基に、Sytl1やそのパートナーであるRab27の機能を抑える工夫を考えることにより、新たな治療薬の開発推進に貢献できることが期待できます。また、Sytl1の発現が高いがんには、白血病の他にも肺の扁平上皮癌がんや一部の乳がんがあることから、がん細胞と周囲の微小環境との相互作用一般に関わっている可能性も考えられます。本研究の成果を通して治療抵抗性の白血病の克服に貢献することが期待できます。
<参考図>
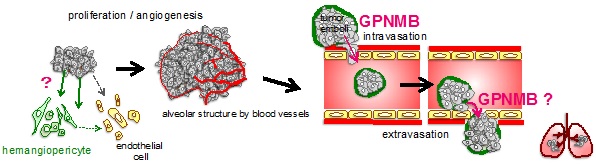
図1:Meis1とSytl1による白血病細胞の骨髄定着を促進する仕組み。左写真は、骨髄内の白血病細胞(緑色)とCAR細胞(赤色)が接している部分。右図は、今回明らかになった分子機構で、Meis1によるSytll1の転写活性化により細胞内小胞に含まれるCXCR4の膜輸送が盛んになり、白血病細胞とCAR細胞の相互作用が促進される仕組みを示している。
<用語解説>
注1)転写因子:
遺伝子DNAからメッセンジャーRNAを産生する転写作用を調節する因子。比較的短いが特異的なDNA配列に結合する領域を有していて、この性質を利用することで自身の標的遺伝子に結合して、さらに転写共役因子と会合することによって、遺伝子発現を時間的・空間的に調節している。但し、染色体上に存在するDNA配列に結合するに際しては、ヒストンの修飾状態やDNAのメチル化状態などエピゲノムの状況が大きく影響しているため、細胞種によって働きが大きく異なる。
注2)骨髄ニッチ:
成人の造血は骨髄で行われるが、造血幹細胞が正常に機能するためには、間質細胞とのシグナル伝達を介した相互作用が必要である。骨髄の間質細胞の中で、血管内皮細胞や骨芽細胞、間葉系前駆細胞等が造血幹細胞を支持する骨髄ニッチ(造血ニッチ)を形成すると考えられている。特に、本研究グループが注目したCAR細胞は脂肪と骨芽細胞に分化可能な前駆細胞と考えられていて、CXCL12を分泌することが知られている。造血幹細胞は、骨髄ニッチに存在して、自己複製能や多分化能を維持していることが想定されている。白血病細胞は、この骨髄ニッチを占拠して自身の生存・増殖の場としていると考えられている。
注3)サイトカイン
細胞間の情報伝達物質として働く比較的低分子量の分泌タンパク質。各サイトカインは、情報を受け取る側の細胞表面の膜に存在する特異的な受容体と結合して、細胞の分裂、遊走、形態変化、分化、細胞死といった重要な生理活性を誘導する。数多くのサイトカインが知られているが、代表的なものとして、インターロイキン(IL-6など)、ケモカイン(CXCL12など)、細胞増殖因子(EGF、TGF-betaなど)、エリスロポエチンなどが挙げられる。過剰発現によって、がん遺伝子として作用するものや、エリスロポエチンやG-CSF、インターフェロンなど治療薬として用いられているものもある。
<論文名、著者およびその所属>
○論文名
MEIS1-mediated transactivation of synaptotagmin like 1 promotes CXCL12/CXCR4 signaling and luekemogenesis
○ジャーナル名
Journal of Clinical Investigation
○著者
Takashi Yokoyama1, Mayuka Nakatake1, Takeshi Kuwata1,2, Arnaud Couzinet1, Ryo Goitsuka3, Shuichi Tsutsumi4, Hiroyuki Aburatani4, Peter JM Valk5, Ruud Delwel5, Takuro Nakamura1*
* 責任著者(中村 卓郎)
○著者の所属機関
1 がん研究会 がん研究所 発がん研究部
2 国立がん研究センター 東病院 先端医療開発センター
3 東京理科大学 生命医科学研究所 発生及び老化研究部門
4 東京大学 先端科学技術センター ゲノムサイエンス分野
5 エラスムス大学 血液学講座
<本研究への支援>
本研究は、主に下記機関より資金的支援を受けて実施されました。
・文部科学省 科学研究費補助金 基盤研究(A)、基盤研究(C)、若手研究(B)、挑戦的萌芽研究
<お問い合わせ先>
【本研究の内容に関すること】
公益財団法人がん研究会 がん研究所 発がん研究部
中村 卓郎
TEL:03-3570-0462 FAX:03-3570-0463 e-mail: takuro-ind@umin.net
【取材に関すること】
公益財団法人がん研究会 広報部
本山、大関
TEL:03-3520-0111 FAX:03-3520-0141 e-mail: kouhouka@jfcr.or.jp
![]()

