![]()
- HOME
- 新着情報

【ニュースリリース】RET融合遺伝子陽性肺がんの新たな分子標的薬耐性機構を発見 〜MIG6欠損によるEGFRシグナル活性化を介したRET阻害薬耐性〜
2024年09月13日
1.ポイント
・3次元浮遊培養はRET陽性肺がん細胞のRET-阻害薬感受性を再現しました。
・CRISPR/Cas9ノックアウトスクリーニングにより、MIG6の欠損がRET阻害薬耐性細胞を増加させることを明らかにしました。
・MIG6を欠損した肺がん細胞では、ごく微量のEGFによってEGFRが活性化してRET阻害薬に耐性となること、その耐性は抗EGFR抗体の併用で克服できる可能性を示しました。
・RET肺がん患者由来のがん細胞株の一部では、EGFR活性化による初期耐性が認められました。
2.研究の概要
非小細胞肺がんは肺がんの85%程度を占めますが、その中の1〜2%ではRET融合遺伝子(注1)が検出されます。これらの患者さんではRETチロシンキナーゼを標的とした分子標的薬(RET阻害薬:注2)が高い抗腫瘍効果を示すことが知られています。しかし、治療開始当初は腫瘍の大幅な縮小を認めても数年程度経過すると薬剤が効かなくなってしまう薬剤耐性を獲得し、がんは再び増悪・進行してしまうことが問題となっています。
がん研究会の片山量平(がん化学療法センター 基礎研究部 部長)、魏 薪兆(WEI Xinzhao)(同研究部所属、東京大学大学院 新領域創成科学研究科 博士課程 大学院生)らの研究グループは、薬剤耐性の芽となる治療残存細胞(DTP細胞)に関連する遺伝子を同定するために、RET融合遺伝子陽性肺がん患者さんから樹立したがん細胞株を用いて、ヒトのゲノムワイドCRISPR/Cas9スクリーニング(注3)を行いました。その結果、複数の候補遺伝子が同定されましたが、その中でもMED12またはMIG6タンパク質をコードするERRFI1遺伝子をノックアウトすると、RET阻害薬存在下で残存するDTP細胞の数が有意に増加しました。MIG6はEGFRに結合しその活性を抑制している分子であり、MIG6を人為的に欠損させると、わずか1 ng/mlという健常人血中濃度レベルのEGFRリガンド(EGFやTGFa)を処理するだけでもEGFR活性化を誘導し、RET阻害薬への耐性をもたらしました。このMIG6ノックアウトによる耐性は、アファチニブといったEGFR阻害薬やセツキシマブといった抗EGFR阻害抗体をRET阻害薬と併用することで、克服されることが判明しました。また、RET阻害薬による治療を受けていないRET融合遺伝子陽性肺がん患者より樹立されたがん細胞株の1つは、元々RET阻害薬に耐性を示しました(初期耐性)。この細胞においても、EGFRシグナルの活性化が生じており、EGFRをAfatinibなどの処理によって阻害するとRET阻害薬耐性は克服されましたが、この細胞ではMIG6は発現しており、EGFRシグナルを介した耐性には関わっていませんでした。本研究では、RET阻害薬抵抗性に関わるメカニズムを同定すると共にその克服法候補を発見することができており、今後の治療法開発に貢献しうる研究成果であると考えます。
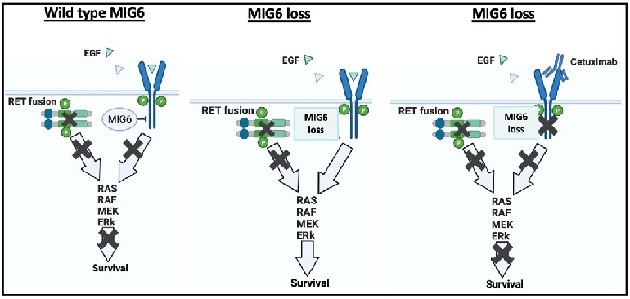
本研究の概要図
MIG6存在下(左)ではEGFRシグナルが抑制されているためRET阻害剤に感受性を示す。MIG6が欠損した細胞(中)ではわずかな量のEGFでもEGFRシグナルが活性化しRET阻害薬耐性が誘導されるため、EGFRを阻害する薬剤や抗EGFR抗体(Cetuximabなど)を併用することで耐性は克服できる(右) BioRenderにより作図
(*注1) RET融合遺伝子
RET(Rearranged during Transfection)遺伝子がコードするRETタンパク質は受容体型チロシンキナーゼです。がん細胞の中には染色体転座や逆位などの現象により、CCDC6やKIF5Bなどの遺伝子とRET遺伝子が融合遺伝子を形成する場合があります。融合遺伝子形成により恒常的にRET融合タンパク質が発現するとともに、多量体形成を介して、恒常的にRETキナーゼが活性化されることでがん化が促進されると考えられています。
(*注2) RET阻害薬
RET融合タンパク質のキナーゼ活性を抑制する分子標的薬です。本邦においては、RET阻害薬としてセルペルカチニブが承認されています。
・3次元浮遊培養はRET陽性肺がん細胞のRET-阻害薬感受性を再現しました。
・CRISPR/Cas9ノックアウトスクリーニングにより、MIG6の欠損がRET阻害薬耐性細胞を増加させることを明らかにしました。
・MIG6を欠損した肺がん細胞では、ごく微量のEGFによってEGFRが活性化してRET阻害薬に耐性となること、その耐性は抗EGFR抗体の併用で克服できる可能性を示しました。
・RET肺がん患者由来のがん細胞株の一部では、EGFR活性化による初期耐性が認められました。
2.研究の概要
非小細胞肺がんは肺がんの85%程度を占めますが、その中の1〜2%ではRET融合遺伝子(注1)が検出されます。これらの患者さんではRETチロシンキナーゼを標的とした分子標的薬(RET阻害薬:注2)が高い抗腫瘍効果を示すことが知られています。しかし、治療開始当初は腫瘍の大幅な縮小を認めても数年程度経過すると薬剤が効かなくなってしまう薬剤耐性を獲得し、がんは再び増悪・進行してしまうことが問題となっています。
がん研究会の片山量平(がん化学療法センター 基礎研究部 部長)、魏 薪兆(WEI Xinzhao)(同研究部所属、東京大学大学院 新領域創成科学研究科 博士課程 大学院生)らの研究グループは、薬剤耐性の芽となる治療残存細胞(DTP細胞)に関連する遺伝子を同定するために、RET融合遺伝子陽性肺がん患者さんから樹立したがん細胞株を用いて、ヒトのゲノムワイドCRISPR/Cas9スクリーニング(注3)を行いました。その結果、複数の候補遺伝子が同定されましたが、その中でもMED12またはMIG6タンパク質をコードするERRFI1遺伝子をノックアウトすると、RET阻害薬存在下で残存するDTP細胞の数が有意に増加しました。MIG6はEGFRに結合しその活性を抑制している分子であり、MIG6を人為的に欠損させると、わずか1 ng/mlという健常人血中濃度レベルのEGFRリガンド(EGFやTGFa)を処理するだけでもEGFR活性化を誘導し、RET阻害薬への耐性をもたらしました。このMIG6ノックアウトによる耐性は、アファチニブといったEGFR阻害薬やセツキシマブといった抗EGFR阻害抗体をRET阻害薬と併用することで、克服されることが判明しました。また、RET阻害薬による治療を受けていないRET融合遺伝子陽性肺がん患者より樹立されたがん細胞株の1つは、元々RET阻害薬に耐性を示しました(初期耐性)。この細胞においても、EGFRシグナルの活性化が生じており、EGFRをAfatinibなどの処理によって阻害するとRET阻害薬耐性は克服されましたが、この細胞ではMIG6は発現しており、EGFRシグナルを介した耐性には関わっていませんでした。本研究では、RET阻害薬抵抗性に関わるメカニズムを同定すると共にその克服法候補を発見することができており、今後の治療法開発に貢献しうる研究成果であると考えます。
本研究の概要図
MIG6存在下(左)ではEGFRシグナルが抑制されているためRET阻害剤に感受性を示す。MIG6が欠損した細胞(中)ではわずかな量のEGFでもEGFRシグナルが活性化しRET阻害薬耐性が誘導されるため、EGFRを阻害する薬剤や抗EGFR抗体(Cetuximabなど)を併用することで耐性は克服できる(右) BioRenderにより作図
(*注1) RET融合遺伝子
RET(Rearranged during Transfection)遺伝子がコードするRETタンパク質は受容体型チロシンキナーゼです。がん細胞の中には染色体転座や逆位などの現象により、CCDC6やKIF5Bなどの遺伝子とRET遺伝子が融合遺伝子を形成する場合があります。融合遺伝子形成により恒常的にRET融合タンパク質が発現するとともに、多量体形成を介して、恒常的にRETキナーゼが活性化されることでがん化が促進されると考えられています。
(*注2) RET阻害薬
RET融合タンパク質のキナーゼ活性を抑制する分子標的薬です。本邦においては、RET阻害薬としてセルペルカチニブが承認されています。
関連PDF
- ニュースリリース文書
 (1,058.8KB )
(1,058.8KB )
![]()